

Issue #62 // Collider Bias In Genomics
Disentangling statistical artifacts from true biological effects


Did you find this piece helpful? If so, tap the 🖤 in the header above. It’s a small gesture that goes a long way in helping me understand what you value and in growing this newsletter. You can also subscribe for free to have the next post delivered to your inbox:
Issue № 62 // Collider Biases In Proteomics Research
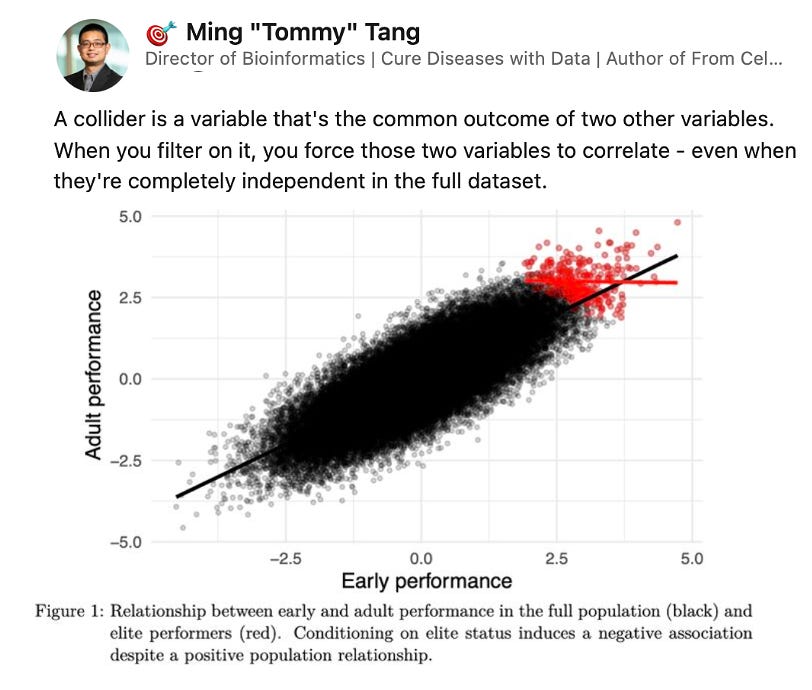
This past week my friend Tommy Tang shared a post on collider biases in genomics research. Collider bias is a statistical distortion that occurs when a variable that is a common effect (a "collider") of two other variables is inadvertently controlled for, creating a spurious association between those two variables. Tommy’s post got me thinking, as it seemed like a potential explanation for something I recently observed in my own research.
First, to give some background, androgen receptor S650, estrogen receptor alpha, and cyclin D1 expression have previously been identified as biomarkers of global treatment resistance among high risk breast cancer patients as reported by Gallagher et al., in Protein signaling and drug target activation signatures to guide therapy prioritization: Therapeutic resistance and sensitivity in the I-SPY 2 Trial. Specifically, when comparing responders versus non-responders in the ISPY-2 trial participant population—where responder is defined by achieving pathologic complete response (pCR) following treatment—these proteins were both up-regulated in non-responders and associated with an increased risk of recurrence (shorted distance recurrence free survival).
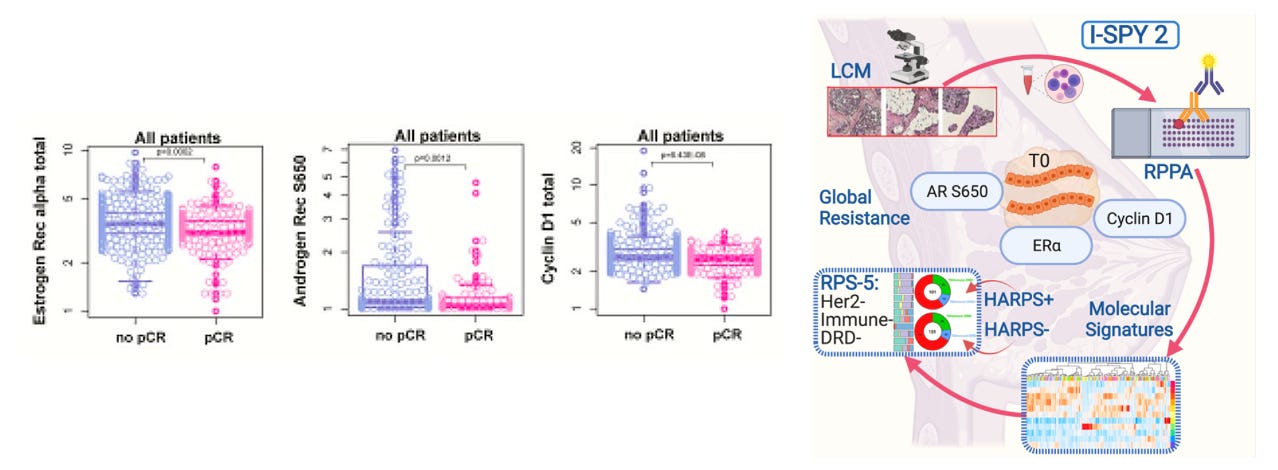
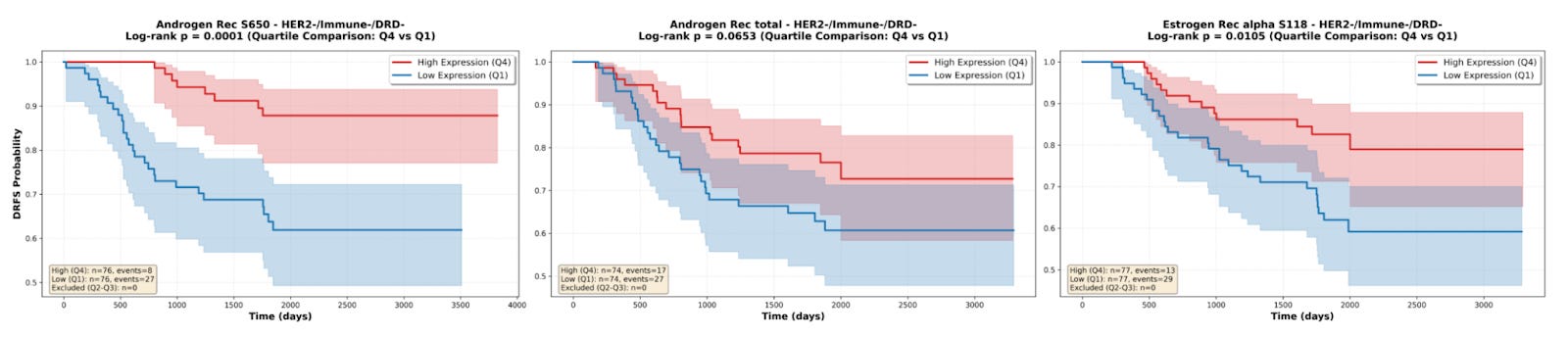
An important note is that despite the ISPY-2 trial being limited to high risk breast cancer, the patient population in the study is fairly heterogeneous, including a mix of patients with HR+/HER2-, HR-/HER2-, HR+/HER2+, and HR-/HER2+ tumors1. Among these HR/HER2 status-based cohorts in the ISPY-2 trial, the HR+/HER2- group had the lowest pCR rate2, and as a result a significant fraction of non-responders in the overall population (approximately ~45%) came from this group. So, it was to my surprise when I was working with reverse phase protein array data from this study, available on the Gene Expression Omnibus via accession number GSE196093, that I found that androgen receptor S650, androgen receptor total, and estrogen receptor alpha S118 were positive prognostic markers in the treatment-refractory HR+/HER2- patient population, with increased expression of all three proteins being associated with a lower risk of recurrence, as demonstrated by the survival curves below.
To make it more explicit, here we have a case where AR and ER are up-regulated in non-responders versus responders and are demonstrated to be negative prognostic markers across then entire patient population (i.e., associated with worse survival outcomes). However, within the non-responder patient population, these same proteins are positive prognostic markers and are associated with more favorable survival outcomes.
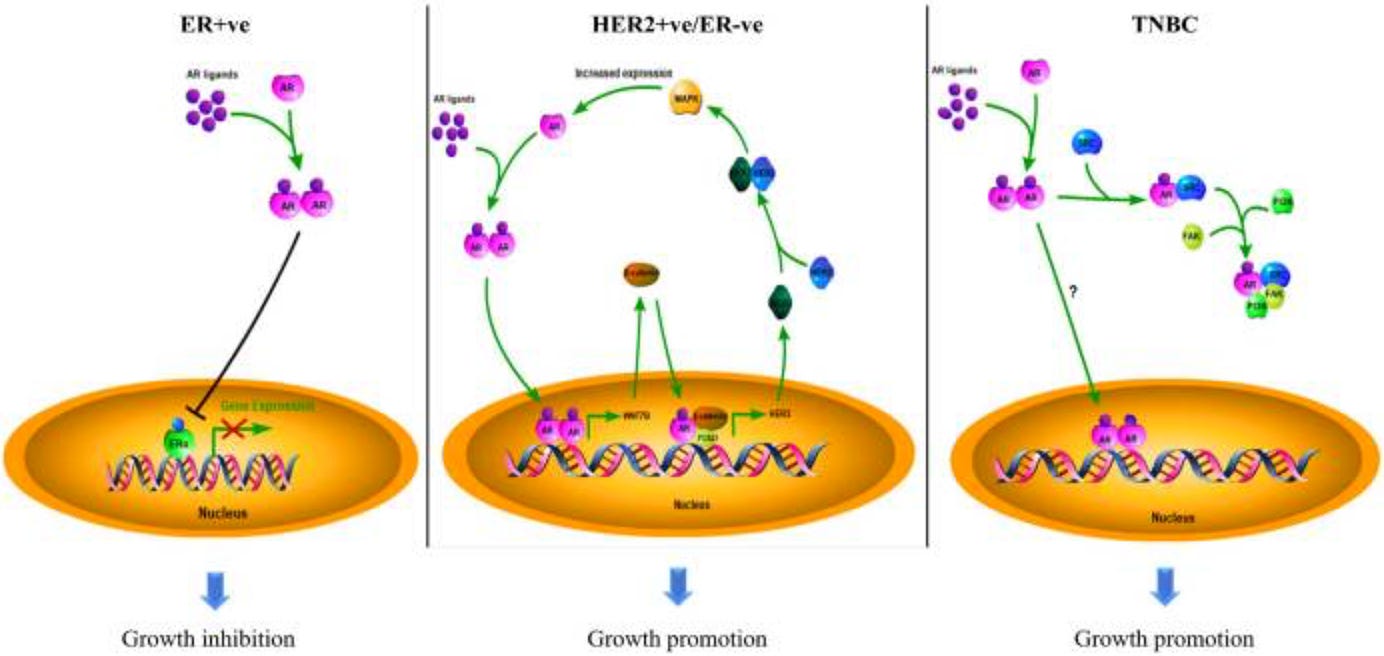
Now, because cancer is a highly heterogeneous disease, we do also need to consider the fact that the prognostic value of androgen and estrogen receptor expression depends on disease context and molecular subtype. Previous survival analyses have shown that AR acts as a tumor suppressor in ER-positive breast cancers, making it a favorable prognostic marker, and a tumor promoter in ER-negative breast cancers, including both ER-/HER2+ and triple negative breast cancer, making it a poor prognostic factor in these contexts (You et al., 2022). This biological heterogeneity could explain part of what we’re observing: the HR+/HER2- non-responders who express higher AR/ER may represent a biologically distinct subgroup where these receptors retain their tumor-suppressive functions despite treatment resistance in this population. This raises an important question: is the observed association in HR+/HER2- non-responders spurious (collider bias) or real, but context-dependent (effect modification)? Can it be both?
The answer to the above question is yes. Collider bias and real effect modification are not mutually exclusive. Collider bias is a statistical structure issue—conditioning on treatment response can induce spurious associations or amplify existing associations. Effect modification, in contrast, reveals a true biological signal. In this case, the causal effect of androgen and estrogen receptor expression genuinely differs by context (breast cancer subtype, tumor stage, treatment arm, etc.), suggesting effect-modification is in play. However, this doesn’t mean that a collider bias isn’t also present. We know that AR/ER have different prognostic effects in hormone receptor positive (HR+) versus hormone receptor negative (HR-) breast cancer, but by conditioning on treatment response (pCR status in this case), we’re selecting a non-random subset of patients. This selection process can still induce or amplify associations beyond what the true biological effect would predict. As a result, even where there’s a real context-dependent biological effect, collider bias can distort the magnitude of effect we observe in the conditioned subset. Thus, the association we observe might be partly real (effect modification) and partly artifactual (collider bias).
This raises the question, how do we determine the degree to which what we’ve observing is collider bias versus real effect? One way to do this is to analyze the protein-outcome association separately within each HR/HER2 subtype in the overall population, then compare these associations with what you observe when you condition on non-responders within each HR/HER2 subtype individually. In my case, I first looked at whether AR/ER are positive or negative prognostic markers within each HR/HER2 subtype population overall. This yielded the following results:
Overall HR+/HER2- population → AR/ER = positive prognostic
Overall HR-/HER2- population → AR/ER = negative prognostic
Overall HR-/HER2+ population → AR/ER = negative prognostic
Overall HR+/HER2+ population → AR/ER = no significant associations
Next, I did a non-responder only analysis within each of these subtypes:
HR+/HER2- Non-responders only → AR/ER = positive prognostic
HR-/HER2- Non-responders only → AR/ER expression is still unfavorable
HR-/HER2+ Non-responders only → AR/ER expression is still unfavorable
HR+/HER2+ Non-responders only → still no significant associations
Notice, the directional associations did not change after conditioning, which suggests that the biological effect we’re observing is real. In other words, androgen and estrogen receptor expression are actually associated with better survival outcomes in the treatment-refractory HR+/HER2- patient population.
To further parse this effect we can also look to see if there is a negative correlation between androgen and estrogen receptor expression and other predictors of non-response, even if these predictors are positively correlated with androgen and estrogen receptor expression in the overall study population. The logic here is that when you condition on a collider (treatment non-response), you create ratification negative correlation between the causes of that collider, even if they are positive or independent in the overall population. Specifically, when conditioning on non-response, we should expect to see that non-responders (patients with high AR/ER expression in this case) will tend to have lower levels of other non-response predictors such as Ki67 levels3. When I tested this with the aforementioned dataset, this does not appear to be the case. Instead, there is no significant association between Ki67 and treatment response in the HR+/HER2- population.
So, back to Tommy’s original post, understanding collider biases and how to disentangle statistical artifacts from true biological signals in an important skill for computational biologists. This is even more so in computational cancer biology, where context-dependence is the norm. We’re constantly faced with scenarios where a given protein acts as a tumor promoter in one context but a suppressor in another, or where a protein signature predicts treatment response in one molecular subtype but non-response in another. As a result, we need to pay careful attention to how studies are designed, what variables are conditioned on (or not), and how our analytical choices may distort true biological signals or introduce spurious associations. The I-SPY 2 dataset is a great testbed for these types of analyses because of its rich omics and clinical data, but we’re rarely so fortunate in practice. In most cases, we lack comprehensive clinical outcome data, detailed patient stratification, or sufficient demographic information. As a result, we need to tread carefully when interpreting our data, assess whether our findings align with the broader literature, and temper our confidence when we lack the data needed to properly disentangle collider bias from effect modification.

HR/HER2 status-based subtypes are themselves heterogenous groups, prompting the creation of response predictive subtypes (RPS) that better predict treatment outcomes than HR/HER2 status alone, as demonstrated in Redefining breast cancer subtypes to guide treatment prioritization and maximize response: Predictive biomarkers across 10 cancer therapies. In this paper Wolf and colleagues identified five RPS subtypes using immune activation signatures, DNA repair deficiency scores, and HER2/luminal phenotypes, resulting in three HER2 negative classifications (HER2-/Immune-/DRD-, HER2-/Immune+, and HER2-/Immune-/DRD+) and two HER2 positive classifications (HER2+/BP-Luminal and HER2+/BP-HER2_or_Basal).
Despite the HR+/HER2- group having the lowest pCR rate, and being associated with a higher residual cancer burden after treatment, the HR-/HER2- (TNBC) group had the shortest distance recurrence free survival.
Ki67 is the percentage of cells that are actively dividing and is therefore used as a proliferation index. A high Ki67 level generally corresponds to a worse prognosis since higher Ki67 indicates a more aggressive cancer. However, because chemotherapies target rapidly dividing cells, high Ki67 often predicts better response to chemotherapy as it reflects a tumor’s innate vulnerability to treatment.