

Issue #77 // Reflections on Cold Spring Harbor Laboratory's AI in Biology Symposium
Have Foundation Models Killed Translational Bioinformatics?


I spent the bulk of last week at Cold Spring Harbor Laboratory’s picturesque campus attending their 90th symposium on AI in Biology where I got to hear talks from world class scientists, including Dr. Jennifer Douda—winner of the 2020 Nobel Prize in Chemistry for her work on the CRISPR-Cas9 genome editing method—among others. My overall impression of the event was that it was equally inspiring and ungrounding, for reasons I’ll elaborate on. By the end of the week the question eating away at me was this: is translational bioinformatics obsolete?

Issue № 77 // Reflections on Cold Spring Harbor Laboratory’s AI in Biology Symposium
As a computational cancer biologist, I’m interested in everything from understanding mechanisms of treatment resistance, uncovering latent therapeutic vulnerabilities that can be mechanistically justified, and predicting which patients will respond to a given treatment regimen. I don’t see these goals— understanding and prediction—as separate. And for most of the history of science, they haven’t been. Yet, modern artificial intelligence (as opposed to Good Old-Fashioned AI) is uncoupling them, severing the corpus callosum that binds these two halves of scientific endeavor.
Consider ESMFold2, presented by Dr. Alex Rives—Head of Science at BioHub—on the first night of the conference. In his talk, Dr. Rives described how ESMFold2 can computationally generate mini-binder designs, which when tested in the wet lab bind their target proteins in a high percent of cases (upwards of ~70%, which is impressive to say the least). Still, there is no legible mechanism, no explanatory model a human scientist can parse and reason from. The same is true for ESM-2 is the foundational language model that ESMFold2 is built atop. These models, while outstanding in their capabilities, do not aid human understanding—instead, they bypass the step where understanding would have occurred, jumping straight from question to answer.
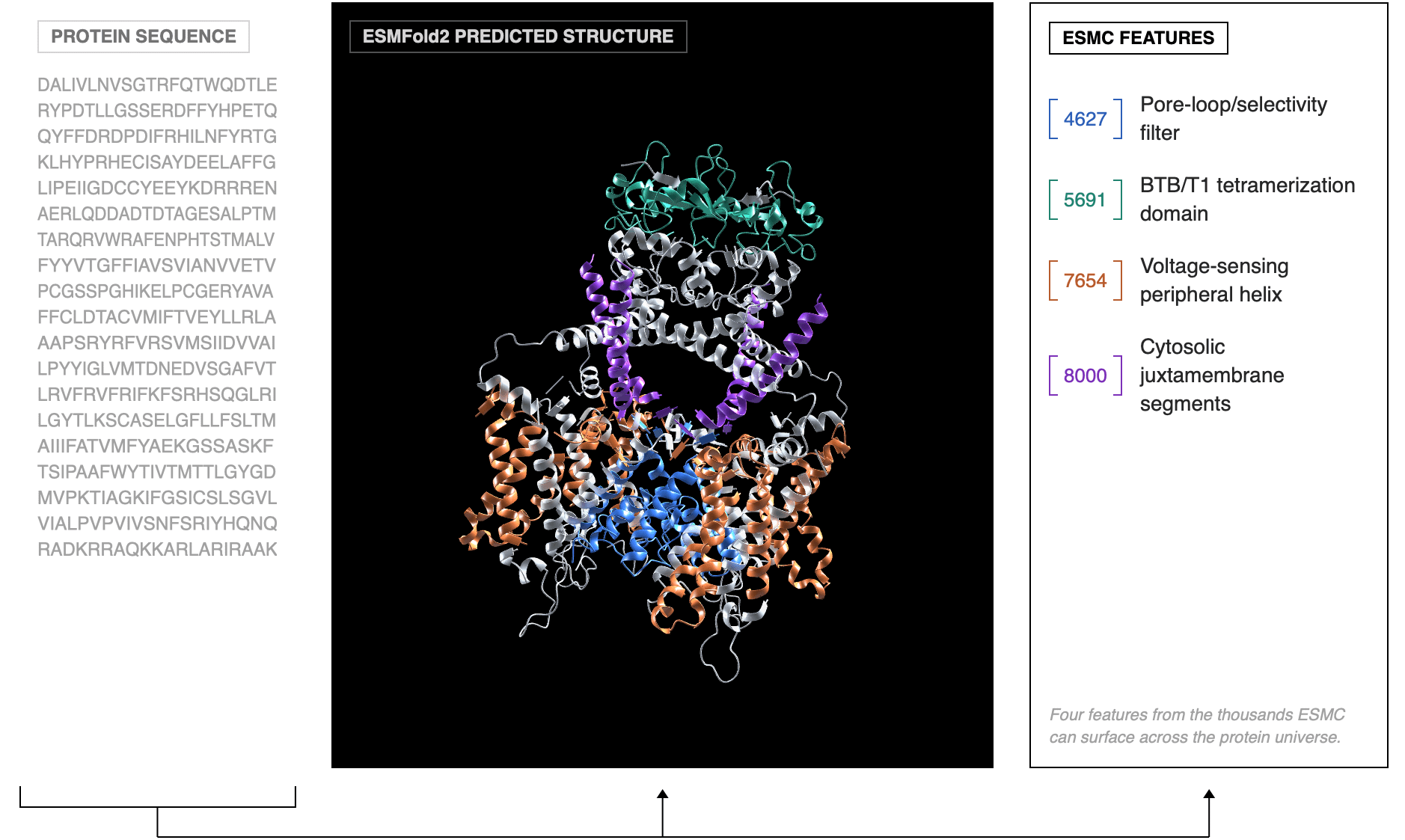
Across a range of talks at Cold Spring Harbor Lab, the cumulative impression I was left with is that this swing in emphasis—from understanding to prediction—is widespread, with different flavors of how this manifests based on the specific research domain. Take the recent wave of foundation models applied to perturbation biology. Companies like Noetik (whose work I find to be extremely cool) are training and tuning large models on spatial genomic, transcriptomic, and proteomics data to predict the downstream effects of genetic and/or chemical perturbations. The implicit bet here is that biological state-space has enough statistical structure that self-supervised pre-training will learn something causally useful—the grammar of biology, so to speak.
Consider what it means for these tools to deliver answers without mechanistic insight, prediction without causal understanding. A good mechanistic model of BCR-ABL kinase signaling easily fits in your head and lets you predict what happens when you add Imatinib (Gleevec)—and crucially, it lets you reason about when resistance emerges when it does. When the T315I gatekeeper mutation appeared clinically, mechanistic understanding of the kinase domain allowed researchers to design second and third generation inhibitors to overcome it, prospectively. A foundation model trained on BCR-ABL perturbations may predict resistance with higher fidelity, but you cannot ask it why in a way that generates a new hypothesis you could test with a different experiment—or use to design the next drug. These tools bypass the step where complex phenomena get distilled into simpler general principles. Without that distillation, we may not be better off than we were before: we couldn’t understand resistance in cancer and now we don’t understand our models of resistance either.
To address this problem, the field of AI mechanistic interpretability was formed, with the explicit goal of shining a light on black-box models and illuminating their inner workings. I find this somewhat ironic; a field dedicated to developing scientific models to understand our scientific models.
Richard Feynman said, “If you can’t explain something to a first-year student, then you haven’t really understood it.” In order to teach someone about a complex set of abstract ideas, you first need to create a mental model. Because a mental model is fundamentally a compression, it easily fits in our head, allowing us to reason about it or simulate it. The capacity to mentally simulate a model is what it means to understand something. AI models have no such constraints. They can hold much more information in their “mind”, which means their internal models can be far less compressed, and far less legible to us by as a result. Put simply, AI models have no need for understanding.

This raises the question, if artificial intelligence generates accurate predictions lead to drugs that effectively kill cancer, does understanding still have a place? I suspect that if predictions and their downstream benefits arrive without understanding, patients will be just fine with that. Yet, there is a strong counter argument to that, which I wrote about previously in Issue 60: Compression as Understanding. In George Church’s words:
“The most powerful sciences are the ones that are better articulated mechanistically on a solid foundation rather than black boxes. The black boxes tend to include artifacts, dead ends. Most of the progress in science and engineering has been part of community efforts with strong mechanistic underpinnings.”
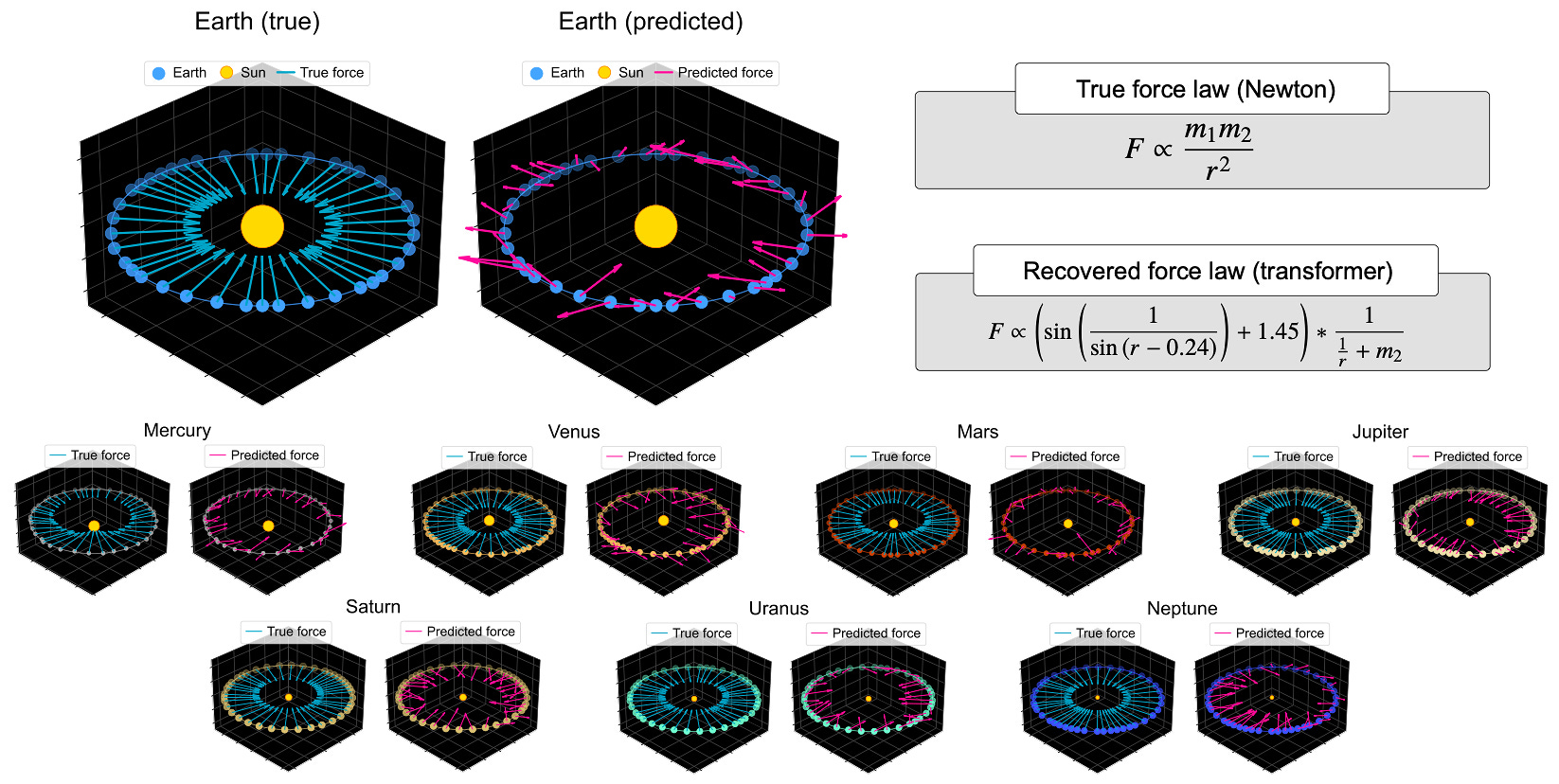
This was sound like sentimentality—nostalgia for a time before AI could make scientific discoveries that humans cannot understand. That said, there is some evidence supporting this stance. Take the paper titled What Has a Foundation Model Found? Using Inductive Bias to Probe for World Models, where a transformer was trained on millions of planetary orbits simulated from Newton’s laws of motion. The model forecasted future positions of simulated planets accurately, but when used to infer the underlying gravitational force vectors it produced a nonsensical force law. Essentially, the transformer had assembled a grab bag of heuristics, accurate for within-distribution predictions, but it hadn’t discovered the universal gravitational principle. Without that principle, the model could predict the movement of solar systems in its training set, but could never land a rover on Mars. This is the cost of prediction without understanding. You can see what will happen. You cannot reason about what to do next.
Now, the question that’s been swirling around in my head since the Cold Spring Harbor Lab symposium is what this all means for computational biologist. Are AIxBIO and translational bioinformatics diverging? Will they continue to circle one another, failing to intersect? Or, will they collide, potentially creating something new? This was a popular dinner conversation, one with marginal consensus, but a lot of interesting opinions. I’m going to do my best to articulate my thoughts on this topic and where I believe there may be an unexplored niche, ripe with opportunity.
A common sentiment expressed by AI researchers at the conference is that they are working on developing a world models of biological processes. This is a very specific bet and applied to foundation models trained on perturbation data it goes something like this: if you expose cells to millions of genetic or chemical perturbations and measure the transcriptomic or proteomic response, the resulting dataset will contain enough statistical structure that an AI model can learn a compressed representation of how biological state space is organized (i.e., what directions in that space correspond to meaningful biology, which perturbations move the system where, and how those effects compose). The hope here is that these learned representations are not arbitrary—that nearby points in embedding space correspond to cells in similar biological states, that directions in the space correspond to interpretable biological programs, and that you can use the geometry of the space to reason about interventions you haven’t tried. This is what people mean when they talk about an AI model learning biological grammar—the latent rules that constrain which states are reachable and how.
This is different from translational bioinformatics, which operates on what we might think of as a feature-centric, marginal inference framework. In essence, this type of framework asks: does this protein’s abundance differ between conditions? Is this difference statistically significant? Does it associate with survival? The logic here is reductive, but in a helpful sense—isolate the signal, validate it, built clinical utility around it. A protein that is upregulated in triple negative breast cancer, whose expression is associated with worse survival, and is druggable is a good candidate target because the chain of inference is short, legible, and most importantly falsifiable.
Now, juxtapose this with AI models, which utilizes a system-level, joint inference framework. The claim here (which I agree with) is that individual features don’t tell you enough because biological systems are highly interdependent—the effect of a perturbation, for example, depends on the state of everything else. A protein that looks like a good target in isolation may be irrelevant in a specific tumor subtype because the network has rewired around it. A model, trained on enough joint observations, should learn these dependencies implicitly.
We can already start to see a superficial divergence: translational bioinformatics produces candidates that are individually interpretable and clinically actionable on short time horizons. AI models produce predictions that are potentially more accurate but harder to interpret, harder to validate, and much more data-hungry. The deeper divergence is in philosophical framing though. Whereas translational bioinformatics assumes biological systems are decomposable—that you can understand parts independently, AI models by contrast implicit assume non-decomposability—that joint structure matters.
Does this make translational bioinformatics obsolete? I think the answer hinges on what prediction without understanding actually costs you—and for certain classes of clinical problems, that cost is high. This inherently pits translational bioinformatics against modern AI frameworks. Reductionism vs holism. Legibility vs illegibility. What’s often neglected though is the third option—that we can recover the joint structure of biological systems without decomposing them into independent parts or handing their complexity to a black box. This is where complex system science comes in.
Both modern AI frameworks and complex systems science see biological systems as inherently non-decomposable, but what they do next is where they differ. AI models treat the cell as a statistical object. In this world view a cell state is a point in a high-dimensional feature space, and your job is to learn a function that maps features to outcomes. The model is judged by predictive accuracy. Interventions are inputs; cellular responses are outputs. The underlying mechanism doesn’t need to be represented—only the input-output relationship matters. This is the implicit ontology of most foundation model work.
Complex systems science on the other hand treats the cell as a dynamical system. This means cell state is a point in a phase space governed by equations of motion, and the system has stable states (attractors) that it returns to after perturbation, unstable states (bifurcation points) where small changes produce large effects, and barriers between attractors that determine how much energy an intervention needs to cause a state transition. Here, the model is judged by whether it captures this geometry correctly.
The practical difference between these two approaches is as follows: a statistical model tells you that perturbation X shifts gene expression in direction Y. A dynamical model tells you why the cell ends up where it does, whether it will stay there, and what combination of perturbations would be needed to cross a barrier into a different stable state. The statistical model can predict; the dynamical model can explain. Naturally, this may seem that I’m trying to pit these two approaches against each other, but in fact I believe they are complementary descriptions of the same system at different levels of abstraction. The statistical model is an empirical approximation to an underlying dynamical reality. The question is whether the approximation is good enough for the clinical question you’re asking—and for many questions it is. For questions about resistance, plasticity, and combination therapy, where the system’s response to sustained intervention matters, the dynamical framing adds something the statistical one cannot.
The bet I’m placing is that the future isn’t all about foundation models and prediction. We’ll realize the value of understanding, which will lead to a swing back to complex systems—much in the same way that mechanistic understandings of the brain are being used to inform developments in AI (again). But more than a swing back, I think we’ll see something new: translational bioinformatics and complex systems science beginning to merge.
These two traditions have historically operated in parallel. Translational bioinformatics asks which features differ between biological conditions, how confident we are, how we avoid false discoveries, and what effect sizes mean clinically. Complex systems science asks a different set of questions: how do biological systems organize themselves, how can that organization be measured quantitatively, how do systems transition between states, and can those transitions be predicted from baseline measurements?
In this way, complex systems science provides an interpretive backbone that grounds the answers translational bioinformatics produces. When a traditional bioinformatics analysis identifies progressive pathway activation across patient stratification layers, the natural next question is which this pattern consistent with an approach to a bifurcation point. Are we watching a system being pushed toward a catastrophic state transition—and if so, can we predict who crosses the threshold and who doesn’t? That reframing changes what the data is telling you. The same differentially expressed genes, the same survival associations, the same effect sizes, but now interpreted within a framework that asks whether the system is approaching an instability, not just whether a feature is statistically significant.
Translational bioinformatics isn’t obsolete. But the field that survives won’t be the one that tries to out-predict ML on ML’s own terms. It will be the one that integrates the interpretive power of complex systems science to ask richer questions of the same data—building models that are not only legible and falsifiable, but that capture something true about how biological systems are organized and how they fail. The goal isn’t to compete with AI. It’s to remain the part of the enterprise that generates understanding, not just answers.
Thanks for reading! If you found this post useful, please consider subscribing or sharing it with a friend. I regularly shared hands-on computational biology techniques, fresh ways to think about tough problems, and perspectives on a range of related topics.
Hey Evan, I was just reading the book "Chaos" by James Gleick and this article dropped into my notifications. I am amazed at some of the parallels you are suggesting and the ideas of dynamic systems explained in the book. Its a really enjoyable read and I would like to suggest it as a bedtime read :)
You are going to have so much fun once finally the monks explanation of reality gets out and we understand chemistry. The biomedical field and biochemistry are just going to explode. Of course every other field is going to explode as well. We will just have an embarrassing amount of progress.