

Issue #64 // Lossy Compression: What RNA-seq Doesn't Tell You About Cellular State
An information-theoretic argument for functional proteomics in research and clinical practice


Liked this piece? If so, tap the 🖤 in the header above. It’s a small gesture that goes a long way in helping me understand what you value and in growing this newsletter. Thanks so much!
Issue № 64 // Lossy Compression: What RNA-seq Doesn’t Tell You About Cellular State
In Manolis Kellis’ course Computational Biology: Genomes, Networks, Evolution, he states, "The most intuitive way to investigate a certain phenotype is to measure the expression levels of functional proteins present at a given time in the cell. However, measuring the concentration of proteins can be difficult, due to their varying locations, modifications, and contexts in which they are found, as well as due to the incompleteness of the proteome. mRNA expression levels, however, are easier to measure, and are often a good approximation. By measuring the mRNA, we analyze regulation at the transcription level, without the added complications of translational regulation and active protein degradation, which simplifies the analysis at the cost of losing information." This notion is easy to lose sight of when doing RNA-seq analysis. But consider what it means to lose information in the Shannon information theoretic sense.
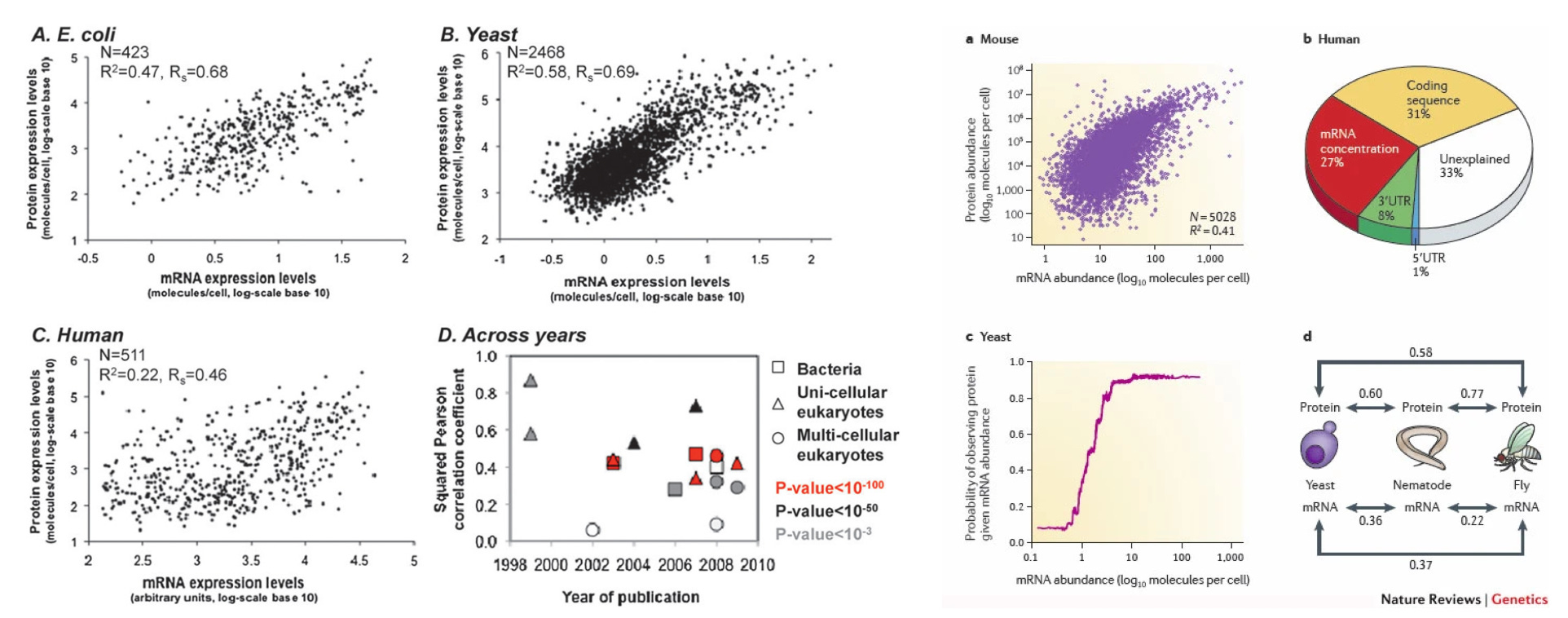
In Claude Shannon’s framework, information is about uncertainty reduction. Each measurement collapses the space of possible states a system could occupy. By stopping at the transcriptional level, we deliberately remain uncertain about an entire regulatory layer. The post-transcriptional processes like alternative splicing, protein modifications, localization, and degradation represent additional sources of information that specify the actual functional state of the cell. The mutual information between mRNA and protein levels is often low to moderate, with correlations around 0.4-0.7, as depicted in the images below, meaning substantial uncertainty remains about the actual effector molecules (as a reference, a correlation of 0.5 between mRNA and protein means ~75% of the variance in protein levels remains unexplained by transcriptomics)1. Some proteins persist for days while their mRNAs turn over in minutes; others are rapidly degraded despite abundant transcripts. Additionally, post-translational modifications can completely alter function without any transcriptional change.
This is where functional proteomics comes in, both as a means of getting closer to a tissue’s phenotype and as a systematic uncertainty reduction tool. Consider HER2-amplified breast cancer. Fluorescence In Situ Hybridization (FISH) can identify gene amplification, giving us a binary answer that reduces uncertainty about genomic state but leaves open whether HER2 drives the tumor’s biology or predicts therapeutic response. Transcriptomics then answers whether HER2 mRNA is elevated, collapsing another degree of freedom, but leaves open the question of whether HER2 protein is abundant. Mass spectrometry proteomics then confirms protein abundance, but functional activity requires something like a reverse phase protein array (RPPA): a high-throughput antibody-based technique that measures protein expression and post-translational modifications, measuring phosphorylated HER2 states. Each layer answers a yes/no question, progressively narrowing the space of possible cellular states until we understand why, within a HER2-amplified population, only a subset responds to targeted therapy.
A simpler way to think about this is as follows: before data collection, we don’t know if a tumor has HER2 gene amplification, whether HER2 gene expression is elevated, or HER2 proteins are abundant and if they are, which sites are phosphorylated, reflecting their activity. Each measurement we take answers one yes or no question. FISH shows HER2 amplification; that’s one point of uncertainty reduced. RNA-seq shows high HER2 expression—we have even less uncertainty about the tissues phenotype. But, it’s not until we move further down the hierarchy that uncertainty is reduced to such a degree that we can make a firm conclusion one way or another2.
The information flow also runs in reverse. Starting from phospho-HER2 elevation and working back through transcriptomics to genomics reveals mechanism—and mechanism determines treatment strategy. High HER2 protein resulting from gene amplification signals oncogene addiction, making pathway inhibition viable. High HER2 protein from impaired degradation, without amplification, suggests the protein may not be a driver, favoring HER2 as a target for antibody-drug conjugates rather than kinase inhibition. The phenotype is identical, but the causal path matters.
Note: Absence of gene amplification doesn’t definitively rule out oncogene addiction—elevated HER2 could still drive proliferation through other mechanisms (e.g., autocrine signaling, receptor heterodimerization). However, it shifts the prior probability: without amplification, HER2 is more likely a passenger or consequence of upstream dysregulation rather than the initiating driver. This probabilistic distinction matters for treatment selection—oncogene addiction predicts durable responses to pathway inhibition, while passenger alterations favor using the protein as a delivery target rather than inhibiting its function. For more on this topic check out Issue #58 // How to Kill a Tumor: Three perspectives on rationale drug selection in personalized cancer therapy.
Understanding this through information theory makes explicit what we’re assuming in our experimental designs. RNA-seq gives us a compressed representation of cellular state—one that is remarkably productive for discovery, suggesting transcriptional regulation captures much of the cell’s control logic. But, the practical convenience of RNA-seq shouldn’t obscure the fundamental gap between what we measure and what actually drives biology. We should recognize when this lossy compression is acceptable and when we need measurements closer to the phenotype itself. In data-driven research, moving down the biological hierarchy reduces uncertainty about what the data mean. In clinical practice, moving up the hierarchy reduces uncertainty about how to intervene. These directions are complementary faces of the same information-theoretic process.
Data-driven research follows the path from genotype to phenotype, asking “what is the cellular state?” to interpret measurements correctly. Clinical practice reverses this, starting from phenotype and tracing back to mechanism, asking “why is this the state?” to select interventions. Both navigate the same multilevel system, but the direction of inquiry determines what uncertainty gets resolved—interpretive uncertainty versus mechanistic uncertainty. Precision medicine requires both: characterizing the actual state (down the hierarchy) and understanding its causal origin (up the hierarchy).
Thanks for reading! If you found this post useful, please consider subscribing. I share hands-on computational biology techniques, fresh ways to think about tough problems, and perspectives on a range of related topics. All free, straight to your inbox.
As Julian Grandvallet-Contreras pointed out here, mRNA/protein correlations for a given gene-protein pair are tissue specific as well, which speaks to varying degree of mutual information between these measurements.
After publishing this piece, I realized that part of my argument may be misleading if taken out of context. The idea that moving from DNA→RNA→protein→phosphoprotein measurements results in progressive uncertainty reduction is only true to the extend that we’re trying to understand the tissues phenotype. However, pretend we live in a universe where phosphoprotein sequencing is the default and DNA sequencing is a rarity. In this world, we may observe elevated HER2 family phosphoproteins in breast tumors, raising the question of whether this result from HER2 amplification. In order to reduce uncertainty in this case we’d move from phosphoprotein/protein→RNA→DNA. As a result, we can’t say that any one -omics measurement is truer that another, as they answer different questions; thus, the mechanics of uncertainty reduction, through orthogonal measurements, is context specific.