Thanks for reading! If you found this post useful, please consider subscribing. I share hands-on computational biology techniques, fresh ways to think about tough problems, and perspectives on a range of related topics. All free, straight to your inbox:
Issue № 68 // Connectivity Is the Target
In the early 2000s, Barabási and colleagues introduced the idea of using network science to identify essential, highly connected protein "hubs" in molecular interaction networks as prime therapeutic targets. The core idea here is that by targeting, and disrupting, these central nodes (which often reside within disease-specific network modules) drugs can collapse pathological signaling while minimizing off target/adverse effects elsewhere.
Conceptually, idea makes a lot of sense to me. But, in practice it’s surprisingly difficult to prove that a given hub is actually a good drug target rather than just a topologically interesting node. This piece is about a framework I’ve been playing around with to do exactly that. I’m writing this partly to clarify my own thinking, and partly to invite feedback from others working on similar problems.
What Makes a Good Drug Target?
In Issue #58: How to Kill a Tumor, I wrote about different schools of thought on drug target identification in cancer. These include finding proteins that are differentially expressed on tumor cell surfaces relative to healthy tissue (the ADC approach), targeting molecular drivers of disease (HER2-directed therapy, AKT inhibitors, and so on), and disrupting network topology, which is the angle Barabási’s work opens up. In an ideal scenario, all three converge on the same protein, resulting in total pathway collapse.
For the first two approaches, the evidentiary chain is relatively clean and easy to understand. Let’s take the case of identifying an ADC (antibody drug conjugate) target, for example. To do this you’d first want to find proteins that are up-regulated in tumors relative to healthy tissue, ensuring that (a) the cytotoxic payload hits the right target and (b) that off-target effects are limited. Next, you’d want to confirm your candidate targets are associated with treatment non-response by seeing if they are further elevated in patients who fail standard-of-care treatment relative to those who response favorable (here, we can define "respond" as achieving pathological complete response, or pCR, for example). Finally, we’d want to show that high expression of the candidate protein predicts disease recurrence in the treatment-refractory population via Kaplan-Meier and Cox proportional hazards analysis. There’s a legible chain of cause and effect here, and the logic maps directly onto clinical intuition—the protein is elevated in tumors, associated with non-response, and patients with high expression of it have worse survival. No one will batt an eye if you use this logic to propose a new drug target.
The network hub case is harder. Many of the standard intuitions about what makes a good drug target don’t cleanly transfer, and even defining what it means for a protein to be a hub is non-trivial. Degree centrality, betweenness centrality, and closeness centrality all capture different aspects of topological importance, and there’s no consensus on which metric is most clinically relevant.
Defining "Core" Hubs
Recently, I’ve been developing the concept of "core hubs," which I define as proteins satisfying two criteria simultaneously1…
First, core hubs must be rich-club members, which are proteins whose degree exceeds the threshold k where the normalized rich-club coefficient (φ_norm) reaches its maximum value in the protein co-expression network. The rich-club phenomenon describes the tendency of nodes of similar degrees to be more densely interconnected with each other than would be expected by chance, forming a kind of elite backbone of the network.
Second, core hubs must also rank in the top 10% of all proteins by degree centrality.
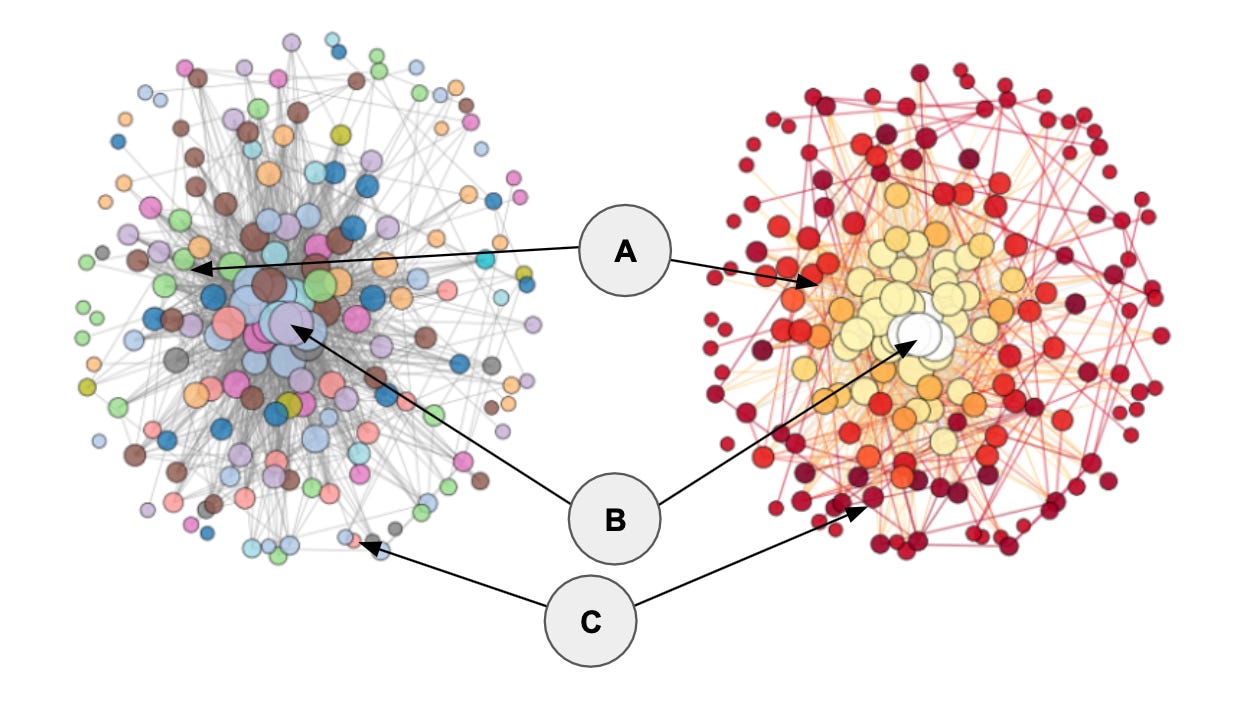
An example: The figure below shows the same protein co-expression network visualized with two complimentary color schemes. The left version is color-coded by pathway membership with nodes sized by their degree centrality. The right version is color-coded by each node’s normalized rich-club coefficient (φ_norm) at their respective degree, with color intensity scaled to the maximum φ_norm.
The node delineated as A is categorized as a core hub, whereas nodes B and C are not. From the visual, you can se that node B has a very high degree centrality (it’s the largest node in the network), but it is not a rich club member (hence, the white color in the rightmost visual). Node C on the other hand is a rich-club member, but it is not highly connected. Only node A, in this comparison, fits both criteria being both highly connected (top 10% of nodes by degree centrally) and a rich-club member.
Conceptually, core hub proteins represent the network's most influential nodes, forming a highly connected, mutually reinforcing, central component that acts as a backbone for efficient global communication. Based on these properties, i’ve hypothesized that disrupting these nodes should cause disproportionate, cascading network failures relative to equivalent disruption of non-hub proteins, making them prime candidates for targeted intervention.
One early observation that gave me some confidence in this framework is how it maps onto established biology. For example, I’ve seen that HER2+ breast cancer cohorts have HER2 family proteins appearing as core hubs in their co-expression networks, which is consistent with the known efficacy of HER2-targeted therapies in these populations and suggests that part of what those therapies are doing is producing network-level collapse rather than simply reducing the abundance of a single target. The framework has also surfaced less expected results: HER2 family core hubs appearing in a subset of TNBC patient networks, and certain ADC target candidates appearing as core hubs in molecular subtypes where you might not anticipate it.
The Validation Problem
The challenge is proving these targets out. Kaplan-Meier curves are less informative when a candidate protein shows limited expression variance across a cohort—which is often the case for network hubs, since their importance is topological rather than purely expression-level. What does become compelling is when a given protein appears as a core hub in a specific molecular subtype (say, TNBC), and then when that population is stratified by treatment response, the hub is specific to the non-responder network. That’s a meaningful signal. But it still doesn’t tell you whether disrupting that hub would actually collapse the network in a therapeutically relevant way.
To address this, I’ve been developing a targeted network attack simulation that works as follows…
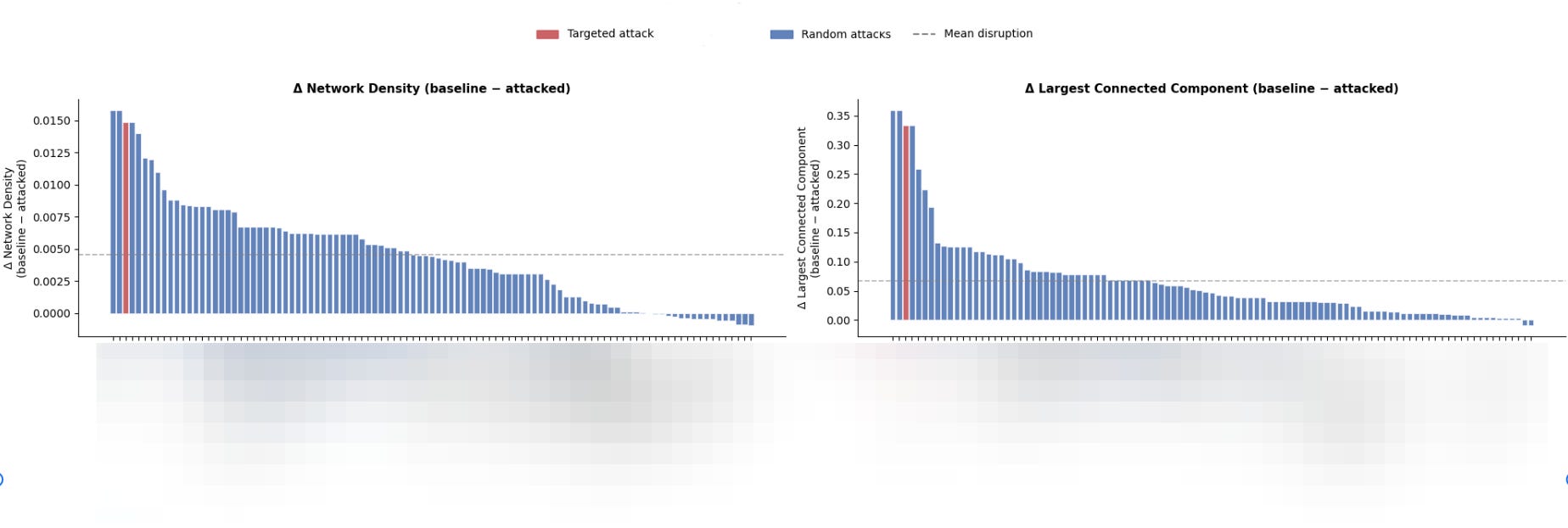
Given a candidate protein A, I simulate a targeted attack by removing that protein and all proteins with co-expression relationships exceeding a defined threshold (|r| > 0.7, for example) from the network.
I then quantify network disruption across four metrics: change in network density, change in the size of the largest connected component, change in average local clustering coefficient, and change in the number of connected components. These metrics collectively capture both global connectivity loss and the degree to which the network fragments into isolated subgraphs.
To contextualize the disruption caused by targeting protein A, I run upwards of 1,000 random attack simulations on the same network, each time selecting proteins at random and removing them along with their high-correlation neighbors. The disruption scores from the targeted attack on protein A are then expressed as a percentile relative to this empirical random attack distribution.
Using the above framework, a core hub should score near the top of this distribution, indicating that its removal causes significantly more network disruption than would be expected from removing a randomly selected protein of equivalent size—evidence that its topological position, not just its connectivity, is what makes it influential.
Outputs from a targeted attack simulation (protein data on the x-axis is redacted). You can see that the core hub candidate protein (red) falls in the 97th percentile of disruption relative to random attacks on the network.
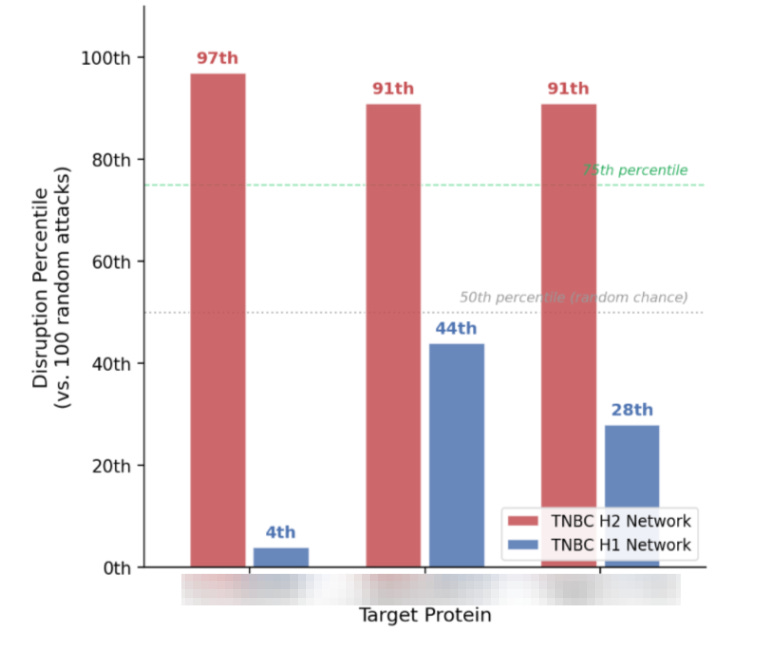
To further validate the target we can then perform a parallel comparison. For example, if the targeted attack is performed on the TNBC non-responder network where the core hub is present, I perform the same attack on the TNBC responder network where the core hub is absent. The prediction is that attacking the hub in non-responders produces near-maximal network disruption, while the same attack in responders produces disruption near the 50th percentile—indistinguishable from random. That divergence would constitute meaningful evidence that the hub’s topological importance is specific to the biological context in which it’s relevant.
An example output from three simulated attacks on MammPrint H1 vs MammaPrint H2 Triple negative breast cancer networks. You can see that attacking these H2-specific core hubs results in maximal disruption in the H2 networks, but not in the comparator H1 networks where these same three candidates fail to achieve score hub status.
One of the most obvious limitations of this approach that I see is that it currently operates on coexpression networks, which capture statistical co-variation between proteins but are agnostic to mechanism. In practice, this could limit some of the causal interpretability of the attack simulations, though I do still think this framework can still meaningfully complement more traditional target validation analyses2.
Additionally, over the next few weeks/months, the extension I’m most interested in is replacing the co-expression networks with causal protein networks, like those generated by CausalEdge. A causal network would allow the attack simulations to operate on directional, mechanistically grounded relationships rather than symmetric correlations, which would substantially strengthen the interpretability of the results. The constraint for now is data: inferring reliable causal networks requires longitudinal proteomic measurements that I don’t currently have at the necessary scale. But it’s a natural next step, and one worth keeping in mind as the framework develops.
Liked this piece? If so, tap the 🖤 in the header above. It’s a small gesture that goes a long way in helping me understand what you value and in growing this newsletter.
I’m not sure if this particular set of criteria has been explored elsewhere (perhaps, under a different name?). if you’ve seen something similar, let me know!
Working with reverse phase protein array (RPPA) data makes this more actionable than it would be with transcriptomic data, for reasons I’ll address in a future piece, but the short version is that RPPA measures protein and phosphoprotein abundance directly, making the co-expression relationships closer to functional reality than mRNA-derived networks would be.