

Issue #74 // A Cluster Isn't A Category
Unsupervised clustering and supervised classification are not analogous, but when combined appropriately can produce outsized effects.


Issue № 74 // A Cluster Is Not A Category
The distinction between classification and clustering is typically introduced early in computational biology and applied ML courses to help differentiate supervised from unsupervised learning. As a general heuristic, classification predicts outcomes, such a therapeutic response, while clustering finds patterns in data. Understanding this distinction is important because these two approaches both require different inputs and answer different biological questions.
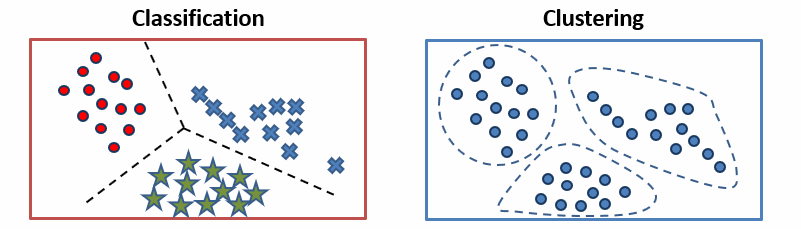
Classification requires training data with labeled examples—such as patient’s protein abundances and treatment outcomes—then learns a decision boundary which is applied to new samples (unseen during training), thus answering the question: “given what we know about these labeled examples, which category does this new sample belong to?” From this problem statement its clear that classification models are appropriate when categories are defined in advance and your goal is to assign new instances to these categories (for example, predicting whether a tumor will response to a drug or calling a variant pathogenic or benign).
Clustering by contrast asks, “what structure exists in our data that we didn’t specify in advance?” When a k-means clustering algorithm groups patients by their gene expression or protein abundance it’s finding underlying patterns without being told what to look for. As a result, clustering is most useful for discover—for example, identifying new candidate genomic subtypes in breast cancer, which can latter be clinically validated. However, we need to be careful with how we interpret clustering results, remembering that a cluster is not a category; it’s a region of high density a particular feature space, defined by a particular distance metric, found by a particular algorithm initialized with particular parameters. Changing any of those will change the clusters.
This distinction is particularly important for computational biologists, as bioinformatics analyses often blur the lines between clustering and classification. Take single-cell RNA-sequencing for example, where unsupervised clustering is used to define cell types and identified clusters are subsequently treated as labels for classification models, resulting in a circular argument. This isn’t always a problem—such as when cell-type clusters are validated by experts or orthogonal measurements prior to being used as labels—but it’s easy to make mistakes here, assuming clusters represent something “real” and concrete when they may just be statistical artifacts (I wrote about this previously in the post below).

Now, that we’ve covered the delineation between clustering and classification, I want to pivot to discussing how these methods can be used synergistically.

Suppose we want to build a classifier to predict the following categorical labels assigned to patients by a molecular oncologist: treatment responder, treatment non-responser, partial responser (stable cases). In this case, let’s assume we have a labeled dataset where index rows are samples (i.e., patients), columns 0-99 are z-score normalized protein abundance values, and the final column contains one of the three aforementioned category labels for each sample. Before we even consider training a classification model, it’s worth asking a prior question: do the labels assigned by the molecular oncologist actually capture real biological variation, or are they arbitrary clinical distinctions imposed on a continuous underlying distribution?
Note: This is exactly the type of question that differentiates someone who is good at ML in computational biology from someone who can run push-button analyses. The separator from good and great practitioners is rarely the code they write—it’s how they interrogate outputs and what they bring to that interrogation that no automated pipeline can replicate.
In order to answer this question, you can run k-means clustering (with the “optimal” number of clusters) on your z-score normalized proteomic features along without touching the category labels. Then, you can use Pandas’ cross-tab function to compare the resulting cluster assignments against patients known response categories. If responders predominantly group in one cluster with non-responders and partial-responders in another, the unsupervised learning model and clinical annotations are well aligned suggesting that the oncologist’s response categories are biologically “real” (i.e., they don’t represent arbitrary boundaries imposed by the human mind).
If on the other hand, responders, non-responders, and partial-responders are distributed across clusters (co-mingling with one another), we have to entertain one of two possibilities: the proteomic features you’ve measured don’t capture the relevant biology or the response categories don’t reflect clean molecular distinctions (or both). In either case, training a classifier would be a waste of time. As a result, we can consider clustering and cross-tabulating to be a sanity check on whether supervised learning (classification) is even warranted.
Assuming our cross-tabulation results look good (i.e., clusters recapitulate classifications), we’re in a good position to build a classifier that can generalize to new, unseen, samples (if you’re interested in learning the details about this process check out How to Develop Predictive Biomarkers). To pull this off we’d take our labeled data—protein features as input, response category as the target—then train a model to learn the mapping that represents a linear or non-linear combination of proteins that best predict treatment response from our labeled examples (training data).
The ability to generalize to new unseen data is one of the qualities that sets classification apart from clustering. A trained classifier learns a decision boundary that can be applied to patients it has never encountered. When we add a new patient’s baseline protein abundance data through a k-means model on the other hand, we’ll get a cluster assignment—importantly, one based on proximity to centroids that shifts when we add new data or alter the distance metric. Clinically, this is the difference between a usable tool and a novel research finding.
In summary, clustering can be used to validate whether label assignments in your data are consistent with biological reality. Classification, by contrast, can be used to build a model that generalizes to new unseen data. The two methods reinforce each other and work synergistically. However, used interchangeably or without understanding what question each one answers, they produce findings that are nonsensical, or misleading.
Thanks for reading! If you found this post useful, please consider subscribing. I share hands-on computational biology techniques, fresh ways to think about tough problems, and perspectives on a range of related topics.